Cell子刊|聚焦7大核心发现,吴爱悯揭示椎间盘退变过程“表观-代谢”互作新机制
| 2026/2/27 10:12:33 《最新论文》 作者:温州医科大学附属第二医院 吴爱悯 我有话说(0人评论) | 字体大小:-│+ |
椎间盘退变(IVDD)是引起慢性腰痛的主要原因之一,全球有近8亿人受其困扰。椎间盘是一个高度结构化的组织,其核心部分——髓核(NP)——处于低氧、低营养的特殊微环境中,主要由髓核细胞(NPCs)和细胞外基质(ECM)构成。NPCs的功能状态直接决定了椎间盘的稳态与退化进程。
近年来,代谢紊乱、线粒体功能障碍和氧化应激被广泛认为是推动IVDD进展的关键因素。特别是线粒体作为细胞的“能量工厂”,其质量控制机制(线粒体自噬,Mitophagy)的失调会导致活性氧(ROS)累积,进而引发细胞衰老和凋亡。然而,NPCs在退化过程中如何维持“代谢-自噬”平衡,其背后的表观遗传分子调控网络仍不清晰。α-酮戊二酸(α-KG)是三羧酸循环(TCA)中的关键代谢中间物,不仅参与能量代谢,还具有抗氧化、抗炎和延缓细胞衰老的多重功能。
研究表明,人体血清中的α-KG水平随年龄增长逐渐下降,而外源性补充α-KG在多种衰老模型中显示出改善组织功能、延长健康寿命的潜力。那么,α-KG的缺失是否是椎间盘退变的“至暗时刻”?补充它能否点亮NPCs的线粒体自噬通路?
基于团队在椎间盘退变与RNA表观遗传调控领域的多年积累,温州医科大学附属第二医院吴爱悯教授团队在Cell Reports上发表了题为:“METTL3 Regulates α-KG-Dependent Mitophagy and Apoptosis in Nucleus Pulposus Cells through the MALAT1/miR-23c/IDH1 Axis”的研究论文。该研究系统描绘了IVDD中的“代谢-表观”交叉调控图谱,揭示了METTL3介导的m6A修饰通过“MALAT1/miR-23c/IDH1”轴调控α-KG合成,进而激活PINK1/Parkin依赖性线粒体自噬的分子机制,为椎间盘退变的防治提供了基于代谢重编程的新策略。
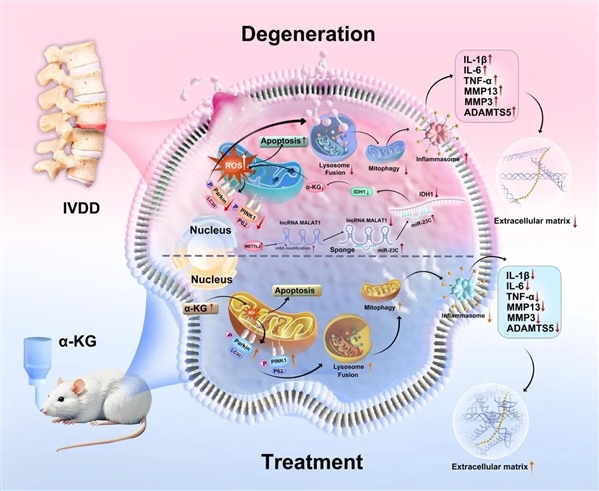
图1:机制示意图
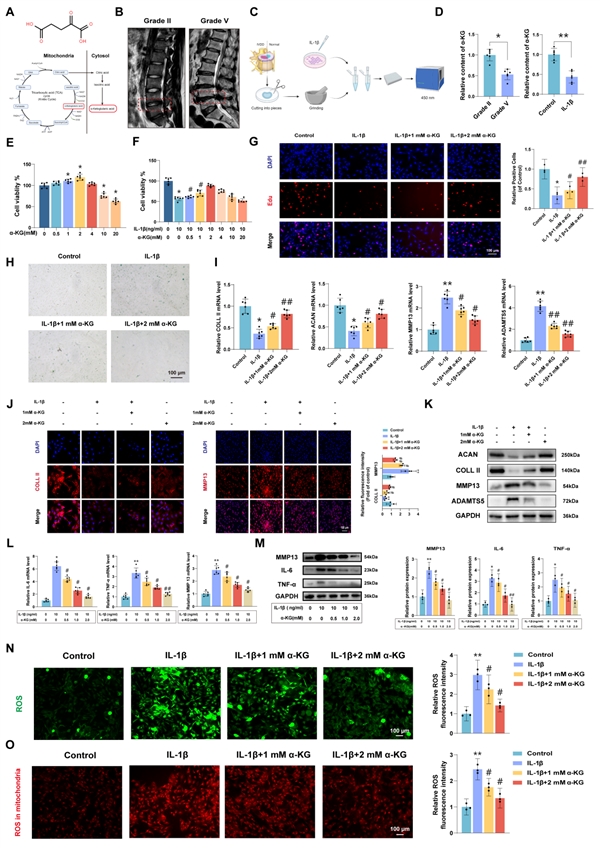
核心发现1:α-KG水平在退变椎间盘组织中显著降低,其补充能逆转退变表型
研究首先发现,在退变程度越高的人椎间盘髓核组织中,α-KG含量越低;体外用炎症因子IL-1β刺激髓核细胞也重现了α-KG的下降。补充α-KG不仅能恢复细胞增殖活力、延缓衰老,还能重塑细胞外基质代谢平衡——促进胶原合成,同时抑制基质降解酶的表达。这提示α-KG减少可能是椎间盘退变的一个关键代谢特征,而外源补充具有治疗潜力。
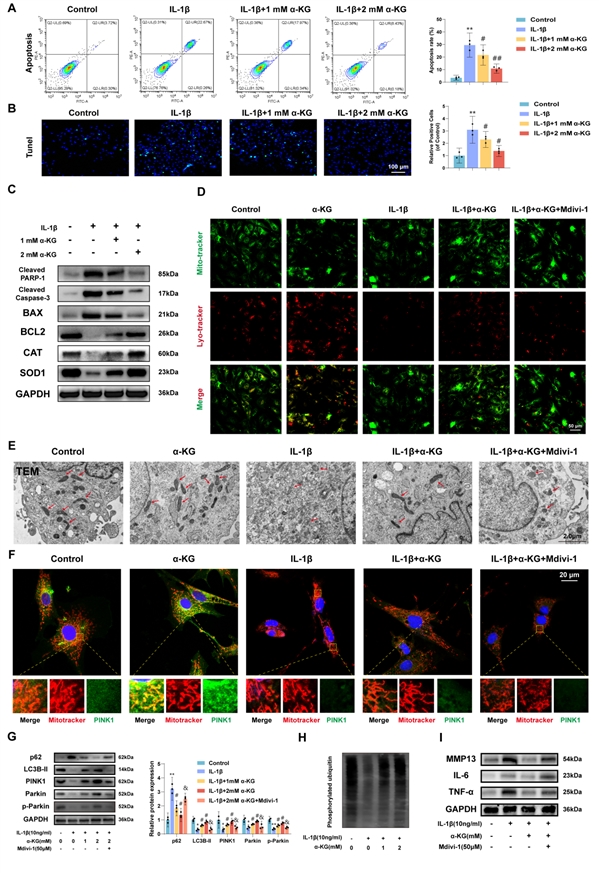
核心发现2:α-KG通过激活线粒体自噬清除ROS,从而抑制细胞凋亡
机制上,α-KG能显著降低IL-1β诱导的活性氧积累,减轻氧化应激损伤。进一步研究发现,α-KG促进了受损线粒体的选择性清除(即线粒体自噬),表现为PINK1/Parkin通路激活及自噬标志物LC3B-II增加。当使用线粒体自噬抑制剂Mdivi-1时,α-KG的抗氧化和抗凋亡作用被完全阻断,这直接证明了α-KG的保护效应依赖于其促进线粒体自噬的能力。
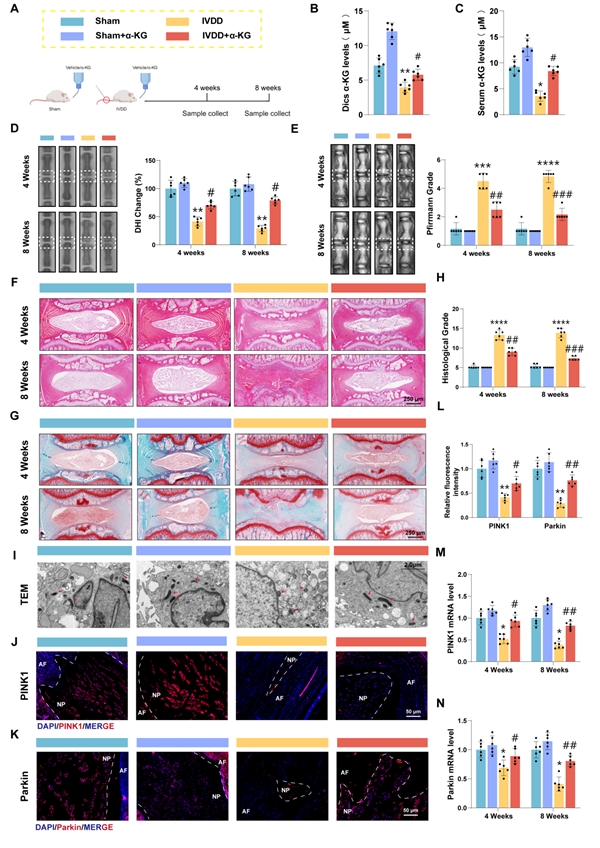
核心发现3:在动物模型中,口服α-KG能通过增强线粒体自噬延缓椎间盘退变进展
在小鼠尾椎穿刺诱导的椎间盘退变模型中,饮用水中补充α-KG能有效恢复髓核内的α-KG水平。影像学和组织学分析显示,α-KG治疗组椎间盘高度保留更好、退变评分更低,髓核结构更完整。同时,退变组织中下调的线粒体自噬关键蛋白PINK1和Parkin得以恢复,电镜下可见线粒体形态改善,从体内证实了α-KG-线粒体自噬轴的治疗作用。
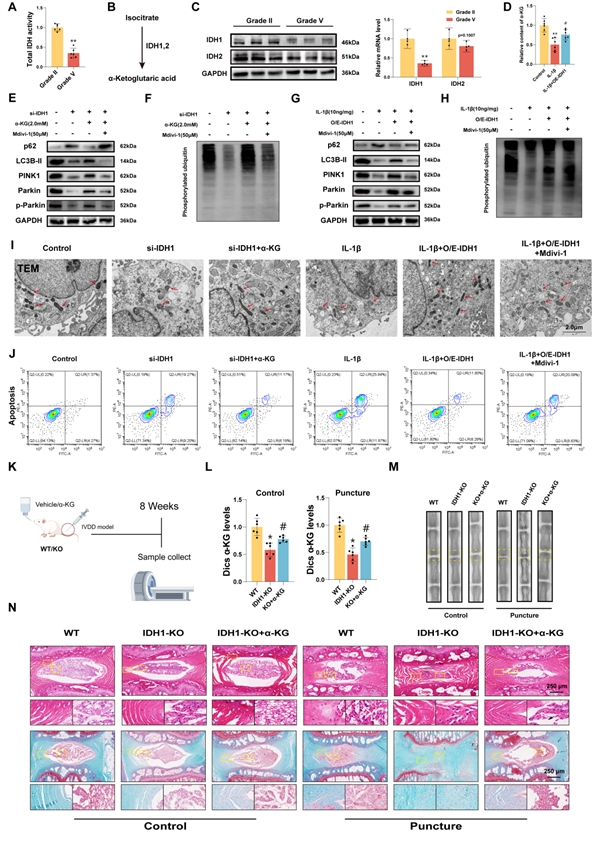
核心发现4:代谢酶IDH1是α-KG生成的关键,其表达下调导致α-KG缺乏与线粒体功能障碍
α-KG从何而来?研究将目光投向三羧酸循环中的关键酶——异柠檬酸脱氢酶(IDH)。发现IDH1在退变髓核组织中表达显著降低,且其酶活性下降。在细胞中敲低IDH1会减少α-KG生成,并重现线粒体自噬抑制、ROS积累和凋亡增加的表型;而过表达IDH1则能逆转IL-1β的损伤。IDH1基因敲除小鼠更易发生椎间盘退变,且对α-KG治疗敏感,确立了IDH1在代谢供应端的核心地位。α-KG从何而来?研究将目光投向三羧酸循环中的关键酶——异柠檬酸脱氢酶(IDH)。发现IDH1在退变髓核组织中表达显著降低,且其酶活性下降。在细胞中敲低IDH1会减少α-KG生成,并重现线粒体自噬抑制、ROS积累和凋亡增加的表型;而过表达IDH1则能逆转IL-1β的损伤。IDH1基因敲除小鼠更易发生椎间盘退变,且对α-KG治疗敏感,确立了IDH1在代谢供应端的核心地位。
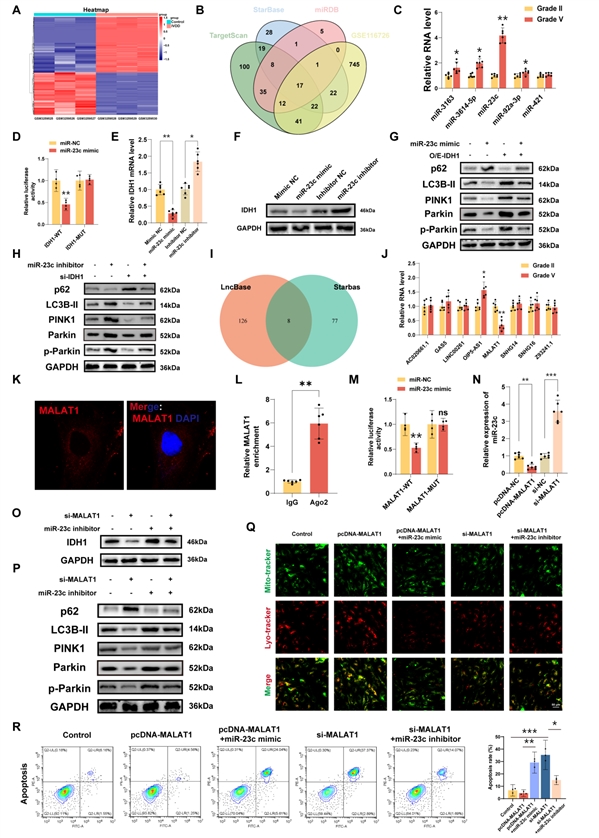
核心发现5:lncRNA MALAT1作为“分子海绵”吸附miR-23c,解除其对IDH1 mRNA的抑制
上游是什么在调控IDH1?通过生物信息学筛选和实验验证,发现miR-23c可直接靶向结合IDH1 mRNA并抑制其翻译。而长链非编码RNA MALAT1在胞质中富集,其转录本上含有miR-23c的结合位点,可像“海绵”一样竞争性结合miR-23c,从而“解放”IDH1 mRNA。过表达MALAT1能上调IDH1、促进线粒体自噬并减少凋亡,而这一效果可被额外添加的miR-23c模拟物所抵消,清晰地展示了ceRNA(竞争性内源RNA)的动态调控网络。
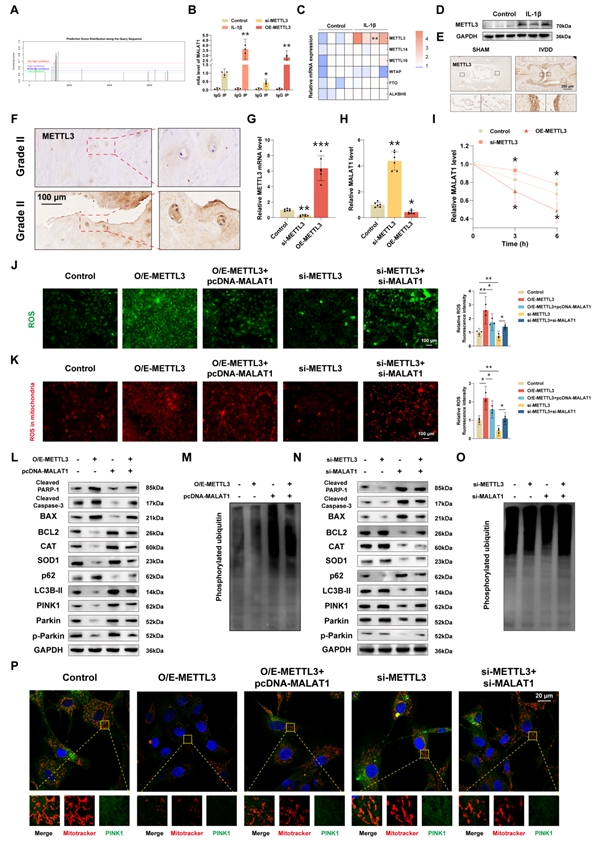
核心发现6:甲基转移酶METTL3介导的m6A修饰负调控MALAT1稳定性,构成上游开关
什么控制了“海绵”MALAT1的丰度?研究表明,在退变环境中,RNA m6A甲基转移酶METTL3表达上调。METTL3在MALAT1转录本上催化形成m6A修饰,此修饰导致MALAT1 RNA稳定性下降、加速降解。过表达METTL3会降低MALAT1水平,进而引发miR-23c上升、IDH1下降的连锁反应,最终抑制线粒体自噬;敲低METTL3则产生相反效果。这揭示了疾病中MALAT1下调的表观转录组学根源。
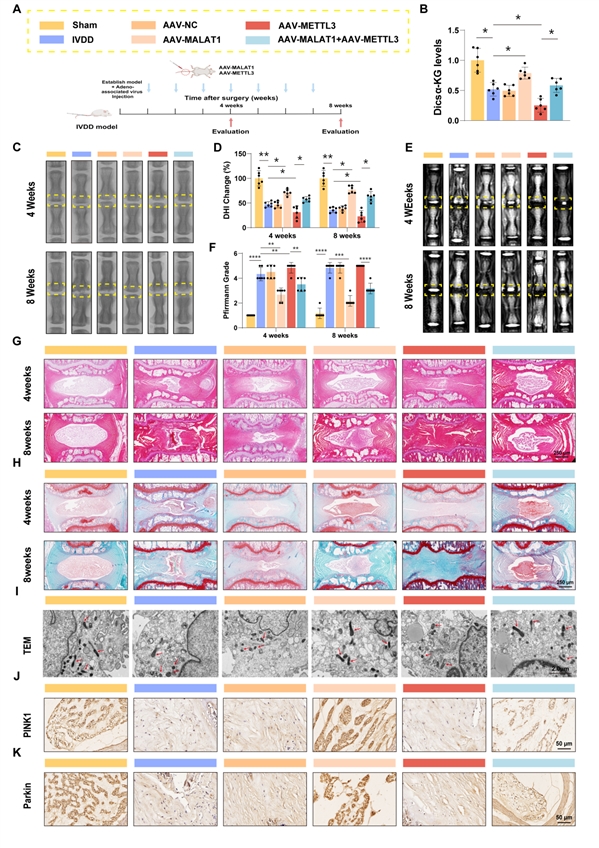
核心发现7:体内干预METTL3/MALAT1轴可有效调控椎间盘退变进程,验证整体通路
最后,研究在动物模型中进行功能挽救实验。向退变椎间盘内注射过表达MALAT1的病毒,能有效提升局部α-KG水平、增强线粒体自噬,并显著延缓椎间盘结构破坏。然而,若同时过表达METTL3,则会抵消MALAT1的治疗获益。这从整体动物水平验证了 “METTL3 ↑ → m6A-MALAT1 ↓ → miR-23c ↑ → IDH1 ↓ → α-KG ↓ → 线粒体自噬抑制 → 椎间盘退变” 这一全新调控轴的存在及其病理重要性。
总结与展望
该研究首次系统阐明了从METTL3介导的m6A修饰,经MALAT1/miR-23c/IDH1信号轴,调控α-KG依赖性线粒体自噬与细胞凋亡的分子通路,不仅深化了对椎间盘退变机制的理解,也为未来开发以α-KG或其上游调控因子为靶点的干预策略提供了理论依据和实验基础。在人口老龄化加剧的今天,该研究为推动IVDD的代谢干预治疗迈出了关键一步,也为其他与衰老相关的退行性疾病的防治提供了新思路。
作者简介

吴爱悯
温州医科大学附属第二医院副院长,主任医师,教授,博士研究生导师及博士后合作导师。
聚焦脊柱疾病基础和临床研究。同时担任多个国内外学术职务,包括国际华人骨研学会(ICMRS)终身会员、AO Spine青年讲师、国际矫形与创伤外科学会(SICOT)中国部青年委员会常务委员,浙江省医师协会青年医师分会副会长,浙江省医学会骨科学分会委员等。
近年来以第一作者或最后通讯作者在Lancet Rheumatology、Lancet Healthy Longevity、Cell Reports, Bone Research, Advanced Science,Advanced Functional Materials、ACS Nano、Bioactive Materials、Experimental & Molecular Medicine等国际期刊发表英文论文60余篇。H指数为52。浙江省杰出青年基金获得者,先后入选浙江省医药卫生高层次人才—创新人才、浙江省医坛新秀、温州市瓯越英才科技领军人才等,并入选爱思唯尔发布的2023-2025“全球前2%TOP科学家”榜单(骨科学方向)。主持包括国家自然科学基金(3项)、中国博士后科学基金一等资助、浙江省重点研发尖兵领雁,及温州市重大科技攻关项目等十余项科研课题。作为第一发明人,获授权美国发明专利2项、中国发明专利7项,其中2项已实现成果转化。研究成果曾获浙江省科技进步奖二等奖(排名第二)、浙江省医药卫生科技奖一等奖(排名第二)、山东省医学科技奖一等奖(排名第三)、中国康复医学会科学技术奖一等奖(排名第五)及上海市医学科技奖二等奖(排名第七)等荣誉。


